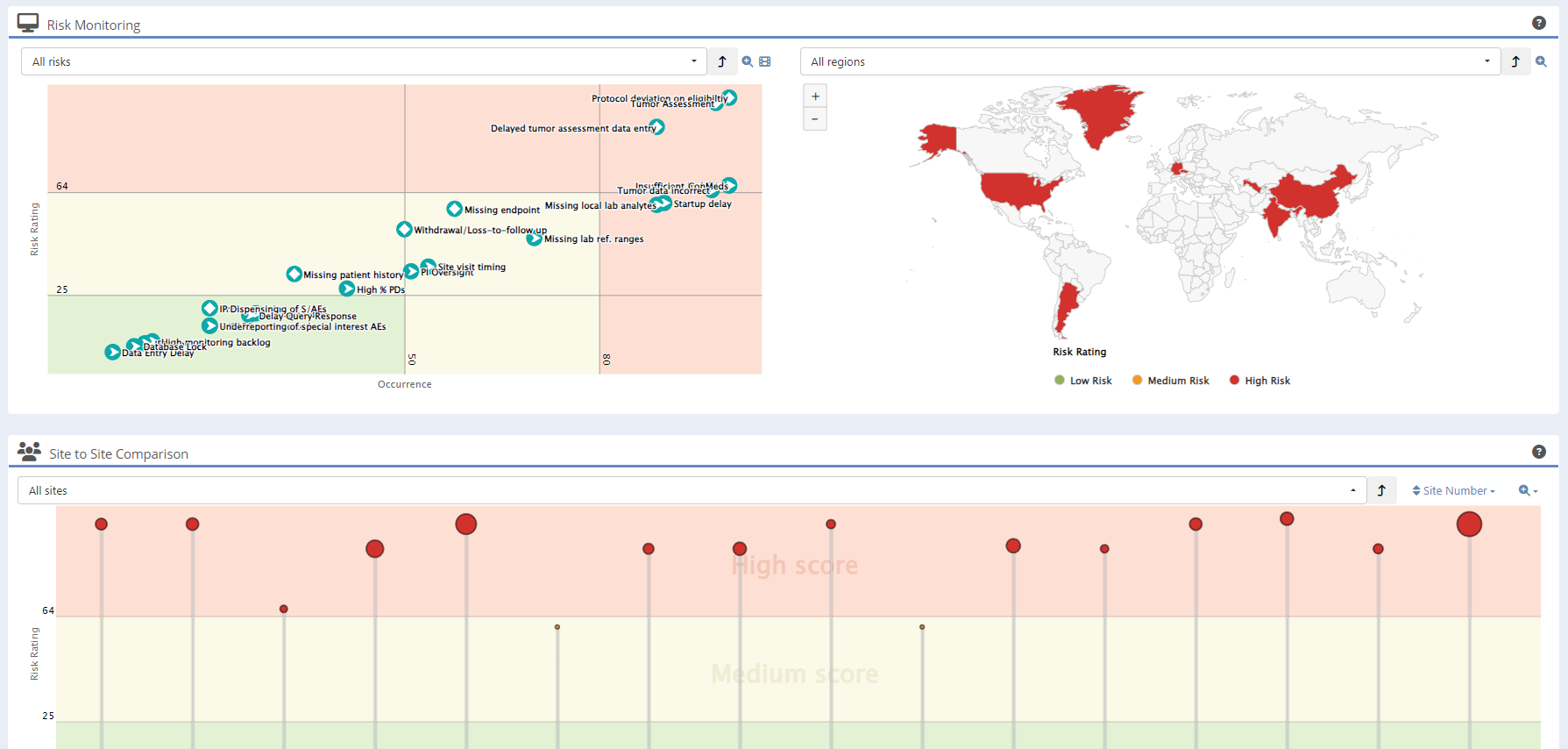
Detect risk patterns early with KRIs, trend shifts, and anomalies
Benchmark regions and sites instantly
Assign, track, and verify corrective actions
See Oversight Come Alive in Real-Time
Dynamic chart interactions, global filters, and drill-downs make it easy to move from signal to action in seconds.
Spot Escalating Issues Before They Harm Your Study
Early visibility of rising risks helps teams act before deviations spread across sites or affect endpoints. Sponsors shorten escalation timelines and reduce downstream cost by knowing where to intervene first.
Understand Where Risk Concentrates Globally
Regional heat-mapping exposes where operational or data-quality issues cluster. This allows monitoring leads to tailor follow-up, allocate CRA effort more efficiently, and maintain compliant oversight across global footprints.
Prioritize the Sites That Need You Most
Side-by-side comparison highlights outliers instantly. Instead of reviewing every site equally, teams concentrate effort on the few that drive most of the risk — improving quality while reducing monitoring workload.
Know Why a Site Is Struggling
Each bar summarizes multiple signals behind a site’s score. Central monitors immediately understand whether issues stem from data delays, eligibility deviations, or workload imbalance.
Filter Out the Noise. Focus on What Matters.
Filter by risk category, severity, or domain to focus reviews. Teams stop spending hours scanning spreadsheets and start making targeted, high-impact decisions.
Unified Global Oversight
Risk signals from all sources converge here: deviations, cycle times, KRIs, queries. No more fragmented oversight — just one clear, actionable picture.
Bring Everything Together
One platform for risk, quality, and performance signals.
Unified Signals, Fewer Surprises
KRIs, deviations, timelines, and quality trends land in one place instead of five different tools. Teams see the same picture, reduce conflicting narratives, and align faster.
From Data Chasing to Decision-Making
Instead of spending days reconciling exports from EDC, CTMS, labs, and vendors, central monitors work from a single, live view. That cuts manual prep and frees capacity for real oversight and follow-up.
One Platform to Show Inspectors How You Stayed in Control
Signals, escalations, and actions are captured in the same system. When QA or inspectors ask how you managed risk, you don’t have to reconstruct the story from scattered files, it’s already there, time-stamped and consistent.
Design out avoidable risk before FPFV. AI-augmented protocol analysis surfaces CtQs, quantifies complexity, predicts deviation hot-spots, and reduces amendment drag, so you lock in patient-safe, inspection-ready design from day one.
Move beyond manual cleaning and fragmented reviews. With time-series anomaly detection, protocol-aware deviation checks, and structured reasoning, the MyRBQM® Portal highlights what truly requires attention.
Stay ahead of issues, not behind them. Portfolio-wide, AI-enhanced dashboards fuse KRIs, QTLs, and deviation signals to cut escalation latency and focus monitors on high-value actions, fully aligned with ICH expectations.
See the full patient story—fast. Subject Profiles synthesize visits, AEs, ConMeds, labs, and trendlines, accelerating case review and improving safety decisions with transparent traceability and RBAC-controlled access.
Our platform and processes meet internationally recognized security, availability, and confidentiality standards. ISO 27001:2022 certification and SOC 2 examinations provide independently verified assurance of our information security controls.
Our clinical-grade AI is independently certified through Microsoft’s Healthcare AI program, ensuring transparent, human-guided oversight, responsible AI practices, and secure integration with Azure’s validated infrastructure.
The MyRBQM Portal is built in alignment with 21 CFR Part 11 and GAMP 5, ensuring data integrity, traceability, and suitability for use in regulated clinical research environments.